Why is PKD1 important and why is it challenging to analyze?
Mutations in PKD1 cause autosomal dominant polycystic kidney disease type 1 (ADPKD1; OMIM *601313). There are a total of 1273 PKD1 variants classified as definitely pathogenic, highly likely pathogenic or likely pathogenic in the PKD1 Mutation Database (Feb 2018).
There is no mutational hot spot for PKD1 and PKD2, which means variants are usually private, highly variable and spread throughout the entire gene. It is estimated that PKD1 explains 85% and PKD2 15% of ADPKD cases. PKD2-related ADPKD is widely acknowledged to be of milder severity than PKD1-related disease.
The genomic region encompassing exons 1–33 of the PKD1 shares 90-99% sequence homology with six known pseudogenes on chromosome 16, making this region very difficult to resolve using traditional NGS methods and resulting in reduced sensitivity to detect disease-causing variants.
This case report sheds light on what kind of testing was chosen for a 25-year-old patient diagnosed with polycystic kidney disease at 4 years of age. The molecular diagnosis allowed for testing of asymptomatic family members to identify those at risk of developing disease.
Patient information
Patient is a 25-year-old female diagnosed with polycystic kidney disease at 4 years of age. There is a family history of ADPKD in multiple generations.
Genetic testing
A Blueprint Genetics Polycystic Kidney Disease Panel was requested which tests for 10 genes including assessment of non-coding variants. The Polycystic Kidney Disease Panel is ideal for patients suspected to have autosomal dominant or autosomal recessive polycystic kidney disease.
Diagnostic summary
The patient was identified to have a pathogenic heterozygous nonsense variant c.4957C>T, p.(Gln1653*) in exon 15 of PKD1. This variant was confirmed with Sanger sequencing using primers that were manually designed to maximize their PKD1 specificity over several highly homologous pseudogenes.
The variant is rare, causes a premature stop codon and is thus predicted to cause loss of normal protein function either through protein truncation or nonsense-mediated mRNA decay. The variant has been listed as pathogenic in the PKD1 Mutation Database, where it has been reported in four families with ADPKD.
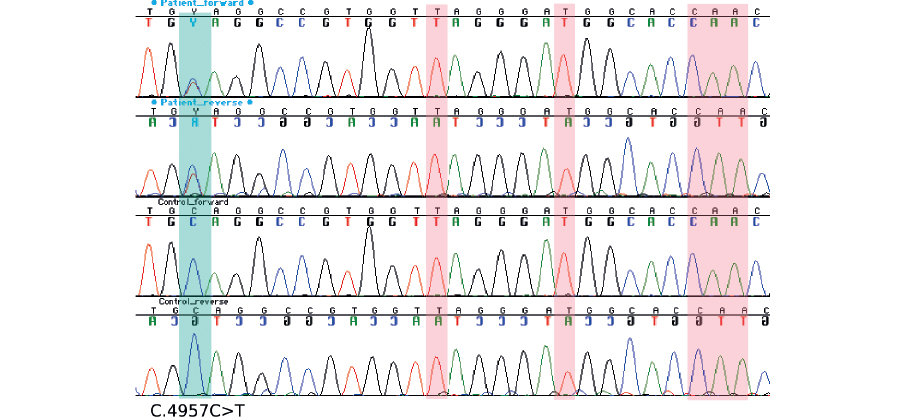
PKD1 has multiple highly homologous pseudogenes in the genome. Patient’s variant c.4957C>T, p.(Gln1653*) is clearly visible in forward and reverse Sanger sequences, while no contaminating signal from the pseudogenes is observed in positions that are unique for PKD1 (marked in red).
Diagnostic implications
- The diagnosis of ADPKD was confirmed.
- Genetic counseling and family member testing are now available to family members to clarify their risk to develop the disease.
- The molecular diagnosis allows for testing of asymptomatic family members to identify those at risk of developing disease in addition to identifying related kidney donors.
Read our white paper The Genetic Horizon – Improving clinical sensitivity in difficult-to-sequence genes for rare hereditary disorders: